22-TA-924
Zāļu ievešanas un izvešanas kārtība
Izdoti saskaņā ar Farmācijas likuma 5.panta 3.punktu un
Narkotisko un psihotropo vielu un zāļu, kā arī prekursoru likumīgās aprites likuma 28.pantu
Narkotisko un psihotropo vielu un zāļu, kā arī prekursoru likumīgās aprites likuma 28.pantu
I.Vispārīgie jautājumi
1.
Noteikumi nosaka:
1.1.
zāļu ievešanas un izvešanas kārtību;
1.2.
muitas kontroles punktus, caur kuriem atļauts Latvijā kontrolējamo narkotisko vielu, psihotropo vielu un prekursoru II sarakstā un III sarakstā iekļauto vielu un zāļu imports un eksports.
1.1
Pētāmo zāļu, kas noteiktas Eiropas Parlamenta un Padomes 2014. gada 16. aprīļa (EK) regulas Nr.536/2014 par cilvēkiem paredzētu zāļu klīniskajām pārbaudēm un ar ko atceļ Direktīvu 2001/20/EK (turpmāk - Eiropas Parlamenta un Padomes regula Nr.536/2014) 2. panta 2. punkta 5. apakšpunktā, (turpmāk - pētāmās zāles) un papildzāļu, kas noteiktas Padomes regulā Nr.536/2014 2.panta 2.punkta 8. apakšpunktā, (turpmāk - papildzāles) importu veic saskaņā ar Eiropas Parlamenta un Padomes 2014. gada 16. aprīļa regulas Nr. 536/2014 IX nodaļu šiem noteikumiem.
2.
Šie noteikumi attiecas uz:
2.1.
to zāļu un papildzāļu (turpmāk - zāles) importu, kuras reģistrētas Latvijas Republikas zāļu reģistrā vai centralizētā reģistrācijas procedūrā saskaņā ar Eiropas Parlamenta un Padomes 2004.gada 31.marta Regulu (EK) Nr. 726/2004, ar ko nosaka cilvēkiem paredzēto un veterināro zāļu reģistrēšanas un uzraudzības Kopienas procedūras un izveido Eiropas Zāļu aģentūru (turpmāk - Eiropas Parlamenta un Padomes regula Nr. 726/2004);
2.2.
to zāļu importu, kuras nav reģistrētas Latvijas Republikas zāļu reģistrā vai centralizētā reģistrācijas procedūrā saskaņā ar Eiropas Parlamenta un Padomes regulu Nr. 726/2004, bet ir reģistrētas trešajās valstīs (turpmāk - nereģistrētas zāles no trešajām valstīm);
2.3.
zāļu importu, ko budžeta iestādes vai sabiedriskā labuma organizācijas vārdā veic zāļu importētājs saskaņā ar Padomes 2009.gada 16.novembra Regulu (EK) Nr. 1186/2009, ar kuru izveido Kopienas sistēmu atbrīvojumiem no muitas nodokļiem (neattiecas uz papildzālēm);
2.4.
zāļu paraugu, tai skaitā vielu, kuras izmanto kā salīdzināmās vielas zāļu testēšanā (turpmāk – standartparaugi), importu;
2.5.
zāļu un zāļu paraugu, tai skaitā standartparaugu, eksportu (izvešanu no Eiropas Savienības muitas teritorijas uz valsti vai tās teritoriju, kas nav Ekonomikas zonas valstis);
2.6.
pētāmo zāļu importu un eksportu.
(Grozīts ar MK 10.06.2008. noteikumiem Nr.415; MK 14.09.2010. noteikumiem Nr.849)
2.1
Brīvostās, speciālajās ekonomiskajās zonās un muitas noliktavās, un citās šo noteikumu 2. punktā minēto zāļu uzglabāšanas vietās zāļu ievešana un izvešana ir pakļauta uzraudzībai saskaņā ar šiem noteikumiem.
3.
Šie noteikumi neattiecas uz:
3.1.
zāļu ievešanu no Eiropas Ekonomikas zonas valstīm un zāļu izvešanu uz Eiropas Ekonomikas zonas valstīm;
3.2.
zāļu personīgai lietošanai ievešanu un izvešanu, ko veic fiziska persona, piemēram, ceļotāja bagāžā;
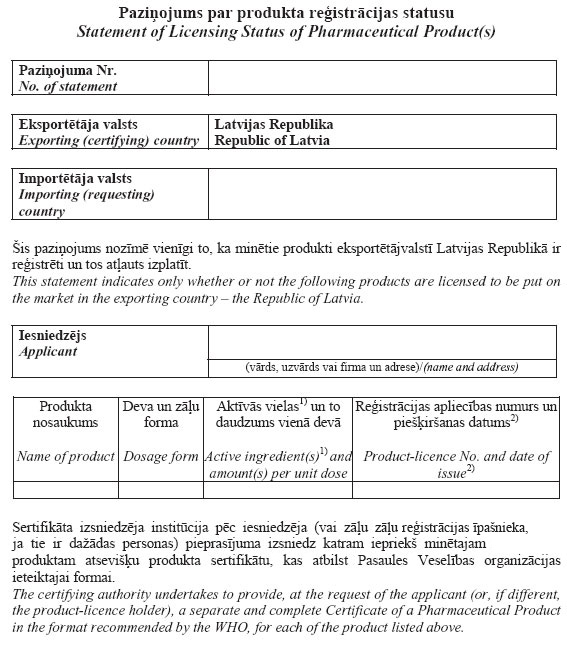
3.3.
zāļu ievešanu un izvešanu, ko veic fiziska persona, zāles saņemot vai nosūtot pasta sūtījumos;
3.4.
veterinārām zālēm.
(Grozīts ar MK 14.09.2010. noteikumiem Nr.849)
4.
Šo noteikumu 2.punktā minēto zāļu (ieskaitot zāles, kuru sastāvā ir Latvijā kontrolējamo narkotisko vielu, psihotropo vielu un prekursoru II sarakstā iekļautās vielas (narkotiskās zāles) un III sarakstā iekļautās vielas (psihotropās zāles)) imports un eksports atļauts caur muitas kontroles punktiem, kuri minēti normatīvajos aktos par valsts robežas šķērsošanas vietu noteikšanu un robežkontroles punktu un robežpārejas punktu izvietojumu uz Latvijas Republikas valsts robežas un kuros atļauts Pārtikas un veterinārā dienesta kontrolei pakļauto nepārtikas preču un produktu imports un eksports.
(Grozīts ar MK 29.09.2009. noteikumiem Nr.1106)
5.
Muitas iestādes kontrolē zāļu importu un eksportu saskaņā ar Muitas likumu un attiecīgajiem normatīvajiem aktiem, kas reglamentē muitošanas un muitas kontroles kārtību.
5.1
Veselības inspekcija veic tirgus uzraudzību attiecībā uz cilvēkiem paredzētajām zālēm un veic tirgus uzraudzības iestādei paredzētos pasākumus atbilstoši prasībām, ko nosaka Eiropas Parlamenta un Padomes 2008. gada 9. jūlija Regula (EK) Nr. 765/2008, ar ko nosaka akreditācijas un tirgus uzraudzības prasības attiecībā uz produktu tirdzniecību un atceļ Regulu (EEK) Nr. 339/93 (turpmāk - Eiropas Parlamenta un Padomes regula Nr. 765/2008).
(MK 02.02.2016. noteikumu Nr. 82 redakcijā)
6.
Pārtikas un veterinārais dienests veic funkcijas saskaņā ar:
6.1.
Eiropas Parlamenta un Padomes regulu Nr. 765/2008;
6.3.
Eiropas Parlamenta un Padomes 2006.gada 17.maija Regulas (EK) Nr. 816/2006 par patentu piespiedu licencēšanu attiecībā uz farmaceitisko produktu ražošanu eksportam uz valstīm, kurās ir sabiedrības veselības aizsardzības problēmas, 14.pantu (turpmāk - Eiropas Parlamenta un Padomes regula Nr. 816/2006).
(Grozīts ar MK 29.09.2009. noteikumiem Nr. 1106; MK 02.02.2016. noteikumiem Nr. 82)
7.
Muitas noliktavai, kurā paredzēts uzglabāt zāles, nepieciešams Veselības inspekcijas atzinums par noliktavas atbilstību zāļu uzglabāšanas prasībām atbilstoši Eiropas Komisijas publicētajām zāļu labas izplatīšanas prakses pamatnostādnēm (valsts valodā pieejamas Zāļu valsts aģentūras tīmekļvietnē).
(MK 29.09.2009. noteikumu Nr. 1106 redakcijā, kas grozīta ar MK 02.02.2016. noteikumiem Nr. 82)
7.1
Veselības inspekcija pārbauda muitas noliktavās, kurā paredzēts uzglabāt zāles ilgāk par 72 stundām telpu, aprīkojuma un iekārtu, un personāla atbilstību normatīvajos aktos par zāļu izplatīšanu noteiktajām prasībām pirms speciālās atļaujas (licences) saņemšanas, kas noteikta normatīvajos aktos par zāļu izplatīšanu, vai pārreģistrējot to. Pēc pārbaudes Veselības inspekcija sagatavo un izsniedz muitas noliktavas atļaujas turētājam pārbaudes aktu, un iesniedz to Zāļu valsts aģentūrā.
7.2
Ja zāles uzglabā muitas noliktavā līdz 72 stundām vai pagaidu uzglabāšanas vietā, attiecīgās muitas noliktavas turētāja un pagaidu uzglabāšanas vietas turētāja pienākums ir informēt Veselības inspekciju par šāda pakalpojuma uzsākšanu pirms tā uzsākšanas.
8.
Zāļu kravas īpašnieks vai valdītājs (turpmāk - zāļu valdītājs):
8.1.
iesniedz muitas noliktavas atļaujas turētājam un pagaidu uzglabāšanas vietas atļaujas turētājam instrukciju, kurā norādītas prasības zāļu uzglabāšanai atbilstoši zāļu ražotāja noteiktajiem uzglabāšanas apstākļiem (zāļu lietošanas instrukcijās, zāļu aprakstos). Muitas noliktavas atļaujas turētājs un pagaidu uzglabāšanas vietas atļaujas turētājs nodrošina zāļu uzglabāšanas apstākļus attiecīgi muitas noliktavā un pagaidu uzglabāšanas vietā saskaņā ar zāļu valdītāja instrukciju un zāļu uzglabāšanas prasībām atbilstoši Eiropas Komisijas publicētajām zāļu labas izplatīšanas prakses pamatnostādnēm (valsts valodā pieejamas Zāļu valsts aģentūras tīmekļvietnē);
8.2.
sedz izdevumus, kas saistīti ar zāļu uzglabāšanas apstākļu nodrošināšanu muitas noliktavā;
8.3.
nodrošina Pārtikas un veterinārā dienesta un Veselības inspekcijas, kā arī muitas iestāžu amatpersonu brīvu pieeju zāļu uzglabāšanas vietai. Zāļu valdītāja atbildīgās amatpersonas pienākums ir uzrādīt zāles Pārtikas un veterinārā dienesta kontrolei.
(Grozīts ar MK 10.06.2008. noteikumiem Nr. 415; MK 29.09.2009. noteikumiem Nr. 1106; MK 14.09.2010. noteikumiem Nr. 849; MK 02.02.2016. noteikumiem Nr. 82)
9.
Zāļu kravai, kuru importē, zāļu valdītājs pievieno attiecīgās ārvalsts izdotu pavaddokumentu, kurā norāda šādu informāciju:
9.1.
zāļu piegādes datumu, zāļu nosaukumu, zāļu formu, zāļu stiprumu vai koncentrāciju un katras piegādātās zāļu ražošanas sērijas numuru un daudzumu, zāļu piegādātāja (nosūtītāja) firmu un adresi, zāļu ražotāja firmu, zāļu ražotājvalsts nosaukumu un zāļu saņēmēja firmu un adresi;
9.2.
cenu, par kādu zāles pārdotas zāļu saņēmējam.
(Grozīts ar MK 13.08.2013. noteikumiem Nr.540)
10.
Ja zāļu valdītājs kravas importam uz līguma pamata izmanto transporta pakalpojumus, ko sniedz cita persona (turpmāk - komercpārvadātājs), tad komercpārvadātājs papildus šo noteikumu 9.punktā noteiktajām prasībām uzrāda muitas iestādē līgumu, kas noslēgts starp zāļu valdītāju un komercpārvadātāju par transporta pakalpojumu sniegšanu, vai zāļu valdītāja pilnvarojumu veikt attiecīgo darbību.
II.Zāļu imports
11.
Zāles drīkst importēt persona, kurai saskaņā ar normatīvajiem aktiem par farmaceitiskās darbības licencēšanas kārtību ir Zāļu valsts aģentūras izsniegta speciāla atļauja (licence) zāļu ražošanai vai importēšanai ar atļauto darbību - zāļu importēšana (neattiecas uz nereģistrētām zālēm, zāļu paraugiem un zāļu tranzītu − zāļu kravu, kuru ieved no trešajām valstīm šo noteikumu 2.1 punktā minētajās vietās un izved uz trešajām valstīm). Pētāmās zāles drīkst importēt persona, kuras speciālajā atļaujā (licencē) zāļu ražošanai/importēšanai norādīts, ka atļauta pētāmo zāļu importēšana. Zāļu kravai, kuru ieved no trešajām valstīm, pamatojoties uz citas Eiropas Savienības dalībvalsts kompetentās iestādes izsniegtu licenci zāļu ražošanai/importēšanai, un transportē cauri Latvijas teritorijai tranzītā (tajā skaitā novieto muitas noliktavā), Zāļu valsts aģentūras izsniegta speciāla atļauja (licence) zāļu ražošanai/importēšanai nav nepieciešama.
(Grozīts ar MK 02.02.2016. noteikumiem Nr. 82)
12.
Persona, kas iesaistīta darbībās, kuru veikšanai nepieciešama šo noteikumu 11.punktā minētā speciālā atļauja (licence) zāļu ražošanai/importēšanai (turpmāk - zāļu importētājs), nodrošina šādu prasību izpildi:
12.1.
importētās zāles, tai skaitā pētāmās zāles, ir ražotas saskaņā ar prasībām, kas ir līdzvērtīgas vai augstākas par Farmācijas likumā un normatīvajos aktos par zāļu ražošanas un kontroles kārtību noteiktajiem labas ražošanas prakses principiem un pamatnostādnēm;
12.2.
zāļu ražotājam attiecīgajā valstī ir atbilstoša atļauja zāļu ražošanai;
12.3.
tās rīcībā pastāvīgi un nepārtraukti ir vismaz viena atbildīgā amatpersona ar atbilstošu izglītību un profesionālo pieredzi (turpmāk - kvalificētā persona). Par kvalificētās personas maiņu nekavējoties, bet ne vēlāk kā piecu dienu laikā rakstiski informē Zāļu valsts aģentūru;
12.4.
tās rīcībā ir personāls, kas atbilst normatīvajos aktos par zāļu ražošanu un kontroli noteiktajām prasībām;
12.5.
nodrošina iespēju Zāļu valsts aģentūras un Veselības inspekcijas amatpersonām jebkurā diennakts laikā apmeklēt zāļu importētāja telpas;
12.7.
importēto zāļu kvalitātes kontrolē un sērijas izlaidē ievēro labas ražošanas prakses principus un pamatnostādnes, kas noteiktas normatīvajos aktos par zāļu ražošanu un kontroli;
12.8.
zāļu izplatīšanā ievēro labas izplatīšanas prakses principus, kas noteikti Eiropas Komisijas publicētajās zāļu labas izplatīšanas prakses pamatnostādnēs (valsts valodā pieejamas Zāļu valsts aģentūras tīmekļvietnē). Attiecībā uz pētāmajām zālēm ievēro normatīvajos aktos par zāļu klīnisko pētījumu veikšanu noteiktās prasības.
(Grozīts ar MK 02.02.2016. noteikumiem Nr. 82; MK 25.09.2018. noteikumiem Nr. 610; 12.2. apakšpunta jaunā redakcija stājas spēkā 31.01.2022., sk. grozījumu 2. punktu)
13.
Kvalificētās personas izglītība un profesionālā pieredze atbilst kvalifikācijas un profesionālās pieredzes kritērijiem, kas noteikti normatīvajos aktos par zāļu ražošanu un kontroli.
14.
Kvalificētā persona, neierobežojot tās attiecības ar zāļu importētāju, ir atbildīga par to, ka katrai importēto zāļu sērijai (arī tad, ja zāles ir ražotas Eiropas Kopienā (Eiropas Savienības un Eiropas Ekonomikas zonas valstis), eksportētas uz trešajām valstīm un importētas atpakaļ) veic pilnu kvalitatīvo analīzi un visu aktīvo vielu kvantitatīvo analīzi, kā arī veic visus citus testus un pārbaudes, kas nepieciešamas, lai nodrošinātu zāļu kvalitāti saskaņā ar zāļu reģistrācijas dokumentācijas prasībām. Zāļu kvalitātes kontroli neveic importēto zāļu sērijām, kuras ir pārbaudītas kādā citā Eiropas Kopienas dalībvalstī, un zāles ir piegādātas no citas dalībvalsts kopā ar kvalificētās personas parakstītu kontroles ziņojumu.
15.
Šo noteikumu 14.punktā minēto zāļu kvalitātes kontroli var neveikt, ja zāles importē no valstīm, kas ar Eiropas Kopienu ir noslēgušas zāļu labas ražošanas prakses atbilstības novērtēšanas savstarpējās atzīšanas līgumu, un šis līgums paredz, ka eksportētājvalstī veic katras zāļu sērijas testēšanu (kvalitatīvo un kvantitatīvo analīzi). Šādā gadījumā katrai importēto zāļu sērijai pievieno šo noteikumu 34.punktā minēto zāļu sērijas sertifikātu.
(Grozīts ar MK 02.02.2016. noteikumiem Nr. 82)
15.1
Šo noteikumu 15. punktā minētais izņēmums piemērojams tikai tām ražošanas darbībām vai zāļu formām, kas norādītas līgumā starp Eiropas Savienību un attiecīgo valsti.
(MK 02.02.2016. noteikumu Nr. 82 redakcijā)
16.
Kvalificētā persona visos gadījumos zāļu sērijas sertificē, izdarot precīzus ierakstus reģistrācijas žurnālā vai citā šim nolūkam paredzētā līdzvērtīgā dokumentā un apliecinot ar parakstu, ka katra zāļu sērija ražota un kontrolēta atbilstoši šo noteikumu 14. un 15.punktā minētajām prasībām. Pēc noteiktām darbībām reģistrācijas žurnālu vai attiecīgo dokumentu papildina un glabā uzņēmumā ne mazāk kā piecus gadus kopš pēdējā ieraksta veikšanas, nodrošinot Zāļu valsts aģentūras un Veselības inspekcijas amatpersonām pieeju minētajam žurnālam vai dokumentam.
(Grozīts ar MK 02.02.2016. noteikumiem Nr. 82)
16.1
Ja zāles ir paredzēts laist tirgū Eiropas Savienībā, kvalificētā persona nodrošina, lai uz attiecīgo zāļu iepakojuma ir zāļu drošuma pazīmes, kas minētas Komisijas 2015. gada 2. oktobra deleģētās Regulas Nr. 2016/161, ar ko papildina Eiropas Parlamenta un Padomes Direktīvu 2001/83/EK, noteicot detalizētus noteikumus par drošuma pazīmēm uz cilvēkiem paredzētu zāļu iesaiņojuma (turpmāk – deleģētā regula Nr. 2016/161), 3. panta 2. punkta "a" un "b" apakšpunktā.
(MK 15.01.2019. noteikumu Nr. 39 redakcijā; punktu piemēro ar 09.02.2019., ievērojot deleģētās regulas Nr. 2016/161 48. un 50. pantā noteiktos pārejas pasākumus, sk. 56.2 punktu)
18.
Līgumā precīzi nosaka pušu pienākumus, īpaši uzsverot līgumdarba izpildītāja pienākumu ievērot labas ražošanas prakses principus un pamatnostādnes, kā arī veidu, kādā kvalificētā persona, kura ir atbildīga par katras sērijas sertificēšanu, izpilda savus pienākumus.
19.
Līgumdarba izpildītājs nodrošina šādu prasību izpildi:
19.1.
ja nav saņemta zāļu importētāja rakstiska atļauja, apakšlīgumu ar trešo personu par darbu, kas saskaņā ar šo noteikumu 17.punktā minēto līgumu uzticēts līgumdarba izpildītājam, neslēdz;
19.2.
ievēro labas ražošanas prakses principus un pamatnostādnes, kas noteiktas normatīvajos aktos par zāļu ražošanu un kontroli, kā arī pakļaujas Zāļu valsts aģentūras kontrolei.
20.
Pirms līguma slēgšanas par zāļu kvalitātes kontroles veikšanu zāļu importētājs nodrošina, ka Zāļu valsts aģentūra sniedz atzinumu par laboratorijas atbilstību labas ražošanas prakses prasībām, kas noteiktas Eiropas Komisijas norādījumos par zāļu un pētāmo zāļu labu ražošanas praksi. Ja laboratorija atrodas citā valstī, Zāļu valsts aģentūra pārliecinās ka konkrētajai laboratorijai Eiropas Savienības datubāzē par ražošanas un importēšanas licencēm un labas ražošanas prakses sertifikātiem (EudraGMDP datubāze) ir spēkā esošs labas ražošanas prakses sertifikāts, kas ietver kvalitātes kontroli attiecīgajam testēšanas veidam, par kuru paredzēts slēgt līgumu.
21.
Attiecībā uz pētāmajām zālēm, kas ražotas trešajās valstīs, kvalificētā persona ir atbildīga par to, ka katra zāļu sērija ir ražota un pārbaudīta saskaņā ar labas ražošanas prakses principiem un pamatnostādnēm (kas ir vismaz līdzvērtīgas Eiropas Savienībā noteiktajām), kā arī saskaņā ar produkta specifikāciju un informāciju, ko sponsors norādījis iesniegumā Zāļu valsts aģentūrai, lai saņemtu atļauju zāļu klīniskai pārbaudei. Pētāmajām zālēm no trešās valsts, kuras klīniskā pārbaudē izmanto salīdzināšanai, un kas ir reģistrētas (atļautas pētāmās zāles), bet par kurām nevar iegūt dokumentāru apliecinājumu, ka katra sērija ir ražota apstākļos, kas ir vismaz līdzvērtīgi labas ražošanas prakses principiem un pamatnostādnēm, kvalificētā persona nodrošina analīžu veikšanu, testus un pārbaudes katrai preparāta sērijai, lai apliecinātu, ka to kvalitāte atbilst informācijai, ko sponsors sniedzis Zāļu valsts aģentūrai, lai saņemtu normatīvajos aktos par zāļu klīniskajiem pētījumiem minēto zāļu klīniskās pārbaudes atļauju vai atļauju ar nosacījumu.
22.
Attiecībā uz pētāmajām zālēm kvalificētā persona visos gadījumos reģistrācijas žurnālā vai citā šim nolūkam paredzētā līdzvērtīgā dokumentā izdara precīzus ierakstus un apliecina ar parakstu, ka katra zāļu sērija atbilst šo noteikumu 21. punkta prasībām. Pēc noteiktām darbībām reģistrācijas žurnālu vai attiecīgo dokumentu papildina un glabā uzņēmumā ne mazāk kā piecus gadus kopš pēdējā pētījuma, kurā tika lietota pētāmo zāļu konkrēta sērija, pabeigšanas vai formālas pārtraukšanas, nodrošinot Zāļu valsts aģentūras amatpersonām pieeju minētajam žurnālam vai dokumentam.
23.
(Svītrots)
24.
Šo noteikumu 11.punktā minētā speciālā atļauja (licence) zāļu ražošanai/importēšanai attiecas tikai uz zālēm (attiecībā uz pētāmajām zālēm - uz zāļu veidiem un zāļu formām), kuras zāļu importētājs ir norādījis iesniegumā speciālas atļaujas (licences) zāļu ražošanai/importēšanai saņemšanai un kuras Zāļu valsts aģentūra, izsniedzot minēto speciālo atļauju (licenci), ir iekļāvusi datu bāzē atbilstoši normatīvajiem aktiem par farmaceitiskās darbības licencēšanas kārtību.
(Grozīts ar MK 02.02.2016. noteikumiem Nr. 82)
25.
Zāļu importētājs drīkst importēt narkotiskās un psihotropās zāles, kuras ir norādītas iesniegumā speciālas atļaujas (licences) zāļu ražošanai vai importēšanai saņemšanai un kuras Zāļu valsts aģentūra, izsniedzot minēto speciālo atļauju (licenci), ir iekļāvusi datubāzē atbilstoši normatīvajiem aktiem par farmaceitiskās darbības licencēšanas kārtību, ja konkrēto zāļu importam saskaņā ar Narkotisko un psihotropo vielu un zāļu, kā arī prekursoru likumīgās aprites likumu ir Zāļu valsts aģentūras izsniegta Apvienoto Nāciju Organizācijas (turpmāk-ANO) Ekonomikas un sociālās padomes Narkotiku komisijas prasībām atbilstoša ikreizēja atļauja. Papildus šajos noteikumos noteiktajām prasībām narkotisko un psihotropo zāļu importēšanā ievēro prasības, kas noteiktas Narkotisko un psihotropo vielu un zāļu, kā arī prekursoru likumīgās aprites likumā.
26.
Zāļu importētājs drīkst importēt budžeta iestādes vai sabiedriskā labuma organizācijas vārdā bezrecepšu zāles, kas iekļautas Latvijas Republikas zāļu reģistrā vai reģistrētas centralizēti saskaņā ar Eiropas Parlamenta un Padomes regulu Nr. 726/2004.
III.Zāļu paraugu un nereģistrētu zāļu imports no trešajām valstīm
27.
Nereģistrētas zāles no trešajām valstīm drīkst importēt persona, kurai saskaņā ar normatīvajiem aktiem par farmaceitiskās darbības licencēšanu ir saņēmusi Zāļu valsts aģentūras izsniegtu speciālu atļauju (licenci) cilvēkiem paredzēto zāļu izplatīšanai vairumtirdzniecībā, kurai ir normatīvajos aktos par zāļu izplatīšanu un kvalitātes kontroli noteiktā kārtībā izsniegta Zāļu valsts aģentūras nereģistrētu zāļu izplatīšanas atļauja individuāli piešķirtām zālēm.
27.1
Nereģistrētas narkotiskās un psihotropās zāles no trešajām valstīm drīkst importēt persona, ja papildus šo noteikumu 27. punktā minētajām prasībām konkrēto zāļu importam saskaņā ar Narkotisko un psihotropo vielu un zāļu, kā arī prekursoru likumīgās aprites likumu ir Zāļu valsts aģentūras izsniegta ANO Ekonomikas un sociālās padomes Narkotiku komisijas prasībām atbilstoša ikreizēja atļauja.
27.2
Prasības, kas noteiktas šo noteikumu 27.punktā, piemēro arī nereģistrētām bezrecepšu zālēm, kas minētas šo noteikumu 26. punktā.
28.
Zāļu paraugus drīkst importēt, pamatojoties uz šo noteikumu 28.1 punktā minēto atļauju, ja tie paredzēti:
28.1.
zāļu reģistrēšanai, iesniedzot tos Zāļu valsts aģentūrā;
28.2.
izmantošanai pētniecībai vai izstrādes pārbaudei Latvijā vai citās valstīs (nav attiecināms uz pētāmām zālēm, tai skaitā zālēm, ko izmanto salīdzināšanai, arī par placebo);
28.3.
izmantošanai mācību vajadzībām;
28.4.
izmantošanai par standartparaugiem testēšanai Latvijā un citās valstīs.
28.1
Zāļu paraugus (izņemot narkotisko un psihotropo zāļu paraugus) no trešajām valstīm drīkst importēt persona, kurai ir Zāļu valsts aģentūras izsniegta atļauja zāļu paraugu importam Latvijas Republikā (1. pielikums). Pēc minētajā atļaujā norādītā zāļu iepakojumu skaita importēšanas atkārtotai zāļu importēšanai nepieciešama jauna atļauja.
(MK 02.02.2016. noteikumu Nr. 82 redakcijā)
28.2
Narkotisko un psihotropo zāļu paraugus drīkst importēt persona, kurai saskaņā ar Narkotisko un psihotropo vielu un zāļu, kā arī prekursoru likumīgās aprites likumu ir Zāļu valsts aģentūras izsniegta ANO Ekonomikas un sociālās padomes Narkotiku komisijas prasībām atbilstoša ikreizēja atļauja. Šajā gadījumā šo noteikumu 28.1punktā minētā Zāļu valsts aģentūras izsniegta atļauja zāļu paraugu importam Latvijas Republikā nav nepieciešama.
29.
Lai saņemtu atļauju zāļu paraugu importam, atļaujas pieprasītājs iesniedz Zāļu valsts aģentūrā iesniegumu atbilstoši šo noteikumu 2.pielikumā noteiktajām prasībām, kurā pamato zāļu paraugu importa nepieciešamību un apliecinājumu par zāļu paraugu izmantošanu tikai konkrētam mērķim.
29.1
Iesniegumu atļaujas zāļu paraugu importam saņemšanai ir tiesīga iesniegt:
29.11.
persona, kura saskaņā ar normatīvajiem aktiem par farmaceitiskās darbības licencēšanas kārtību ir saņēmusi Zāļu valsts aģentūras izsniegtu speciālu atļauju (licenci) zāļu ražošanai vai importēšanai vai speciālu atļauju (licenci) cilvēkiem paredzēto zāļu izplatīšanai vairumtirdzniecībā;
29.12.
Farmācijas likuma 25.1 pantā minētā persona, kurai ir Eiropas Savienības dalībvalstī vai Eiropas Ekonomikas zonas valstī ir izsniegta speciālā atļauja (licence), kas dod tiesības veikt zāļu vairumtirdzniecību vai ražošanu.
29.2
Atļaujā zāļu paraugu importam attiecīgi norāda, kam atļaujas īpašnieks ir tiesīgs izplatīt zāļu paraugus:
29.21.
paraugus zāļu reģistrācijai - personai, kura iesniedz zāles reģistrēšanai Zāļu valsts aģentūrā;
29.22.
paraugus pētniecībai vai izstrādes pārbaudei - personai, kas nodarbojas ar pētniecību vai izstrādes pārbaudi Latvijā vai citās valstīs;
29.23.
paraugus mācību vajadzībām - mācību iestādei;
29.24.
testēšanai - testēšanas laboratorijai Latvijā vai citās valstīs.
30.
Zāļu valsts aģentūra piecu darbdienu laikā pēc šo noteikumu 29.punktā minētā iesnieguma saņemšanas pārbauda, vai sniegtā informācija atbilst šajos noteikumos noteiktajām prasībām. Ja sniegtā informācija ir nepilnīga vai kļūdaina, Zāļu valsts aģentūra rakstiski pieprasa papildu informāciju.
31.
Zāļu valsts aģentūra pieņem lēmumu atteikt izsniegt atļauju zāļu paraugu importam, ja mēneša laikā pēc šo noteikumu 29.punktā minētās papildu informācijas pieprasīšanas nesaņem no iesnieguma iesniedzēja pieprasīto informāciju (pamatojumu).
32.
(Svītrots)
32.1
Izdevumus, kas saistīti ar atļaujas izsniegšanu zāļu paraugu importam, sedz iesnieguma iesniedzējs saskaņā ar Zāļu valsts aģentūras sniegto maksas pakalpojumu cenrādi. Zāļu valsts aģentūra atļauju zāļu paraugu importam atļaujas pieprasītājam izsniedz elektroniska dokumenta formā triju darbdienu laikā pēc lēmuma pieņemšanas, nosūtot to uz iesniegumā norādīto elektroniskā pasta adresi. Papīra dokumenta formā atļauju izsniedz pēc pieprasījuma triju darbdienu laikā par papildu maksu saskaņā ar Zāļu valsts aģentūras sniegto maksas pakalpojumu cenrādi.
IV.Zāļu eksports
33.
Zāles, tai skaitā pētāmās zāles un zāļu paraugus, drīkst eksportēt persona, kura saskaņā ar normatīvajiem aktiem par farmaceitiskās darbības licencēšanas kārtību ir saņēmusi Zāļu valsts aģentūras izsniegtu speciālu atļauju (licenci) zāļu ražošanai vai importēšanai vai speciālu atļauju (licenci) cilvēkiem paredzēto zāļu izplatīšanai vairumtirdzniecībā, kurā ir norādīts speciālās darbības nosacījums – zāļu eksports, vai speciālu atļauju (licenci) zāļu lieltirgotavas atvēršanai (darbībai). Zāļu kravai, kuru izved uz trešajām valstīm, pamatojoties uz citas Eiropas Savienības dalībvalsts kompetentās iestādes izsniegtu licenci zāļu ražošanai/importēšanai, un transportē cauri Latvijas teritorijai tranzītā (tajā skaitā novieto muitas noliktavā), Zāļu valsts aģentūras izsniegta speciāla atļauja (licence) zāļu ražošanai/importēšanai nav nepieciešama.
33.1
Narkotiskās un psihotropās zāles un zāļu paraugus drīkst eksportēt persona, kurai papildus šo noteikumu 33. punktā minētajām speciālajām atļaujām (licencēm) ir konkrēto zāļu eksportam Narkotisko un psihotropo vielu un zāļu, kā arī prekursoru likumīgās aprites likumā noteiktajā kārtībā Zāļu valsts aģentūras izsniegta ANO Ekonomikas un sociālās padomes Narkotiku komisijas prasībām atbilstoša ikreizēja atļauja.
33.2
Persona, kura eksportē zāles, nodrošina, ka:
33.21.
tiek ievērotas Eiropas Komisijas publicētajās zāļu labas izplatīšanas prakses pamatnostādnēs (valsts valodā pieejamas Zāļu valsts aģentūras tīmekļvietnē) noteiktās prasības;
33.22.
zāles piegādā tām personām trešajās valstīs, kuras ir tiesīgas saņemt zāles izplatīšanai vairumtirdzniecībā vai piegādāt tās iedzīvotājiem trešajās valstīs. Visās piegādēs zāļu kravai pievieno dokumentu, kurā ir norādīts:
33.22.1.
piegādes datums;
33.22.2.
zāļu nosaukums, zāļu forma un stiprums vai koncentrācija;
33.22.3.
piegādātais daudzums (par katrām zālēm);
33.22.4.
saņēmēja un piegādātāja nosaukums un adrese;
33.22.5.
katras piegādātās zāļu ražošanas sērijas numurs.
(MK 02.02.2016. noteikumu Nr. 82 redakcijā)
33.3
Persona, kura ir saņēmusi šo noteikumu 28. punktā minētos zāļu paraugus izmantošanai pētniecībai vai izstrādes pārbaudei, vai kā standartparaugus testēšanai, bet šo paraugu izmantošana pētniecībai vai izstrādes pārbaudei, vai testēšanai ir paredzēta trešajā valstī drīkst šo paraugus eksportēt uz attiecīgo valsti izplatīšanai atļaujā norādītajam zāļu paraugu saņēmējam trešajā valstī.
34.
Ja Latvijā reģistrēts zāļu ražotājs eksportē zāles uz valsti, ar kuru Eiropas Kopiena ir noslēgusi zāļu labas ražošanas prakses atbilstības novērtēšanas savstarpējās atzīšanas līgumu, katrai eksportējamo zāļu sērijai pievieno kvalificētās personas parakstītu zāļu sērijas sertifikātu, kurā norādīta informācija saskaņā ar šo noteikumu 3.pielikumu.
35.
Zāļu valsts aģentūra, pamatojoties uz zāļu ražotāja, eksportētāja vai importētājas valsts kompetentās institūcijas iesniegumu, izsniedz:
35.1.
produkta sertifikātu (4.pielikums). Produkts šī punkta izpratnē ir tādas cilvēkiem paredzētās zāles to galīgajā zāļu formā un aktīvās vielas lietošanai šajās zāļu formās, kuras farmāciju reglamentējošajos normatīvajos aktos noteiktajā kārtībā pakļautas kontrolei gan eksportētājvalstī, gan importētājvalstī. Produkta sertifikāts atbilst Pasaules Veselības organizācijas (PVO) ieteiktajai formai un nosaka produkta statusu, kā arī sertifikāta pieprasītāja statusu eksportētājvalstī. Sertifikāts ir paredzēts tikai viena veida produktam;
35.2.
paziņojumu par produkta reģistrācijas statusu (5.pielikums). Šis paziņojums ir paredzēts zāļu importētāja pārstāvim, kurš piedalās starptautiskos piedāvājumos (tenderos) saskaņā ar uzaicinājuma nosacījumiem. Šis paziņojums nozīmē, ka konkrētās zāles Latvijas Republikā (eksportētājvalstī) ir reģistrētas un tās ir atļauts izplatīt. Katram paziņojumā minētajam produktam pēc iesniedzēja un zāļu reģistrācijas īpašnieka, ja tās ir dažādas personas, pieprasījuma Zāļu valsts aģentūra izsniedz šo noteikumu 35.1.apakšpunktā minēto produkta sertifikātu.
(Grozīts ar MK 13.08.2013. noteikumiem Nr.540)
35.1
Zāļu valsts aģentūra, pamatojoties uz zāļu vai aktīvās vielas (produkta) Latvijā reģistrēta ražotāja iesniegumu vai trešās valsts kompetentās institūcijas pieprasījumu, izsniedz produkta sertifikātu saīsinātā formātā – farmaceitiskā produkta sertifikātu vai brīvās tirdzniecības sertifikātu (turpmāk –saīsinātais sertifikāts). Saīsināto sertifikātu izsniedz viena veida produktam (latviešu un angļu valodā). Ja vienām zālēm ir atšķirīgi stiprumi, tās uzskatāmas par viena veida produktu. Ja vienām zālēm ir atšķirīgas zāļu formas (piemēram, tabletes, šķīdums), katrai zāļu formai iesniedz atsevišķu iesniegumu un izsniedz atsevišķu saīsināto sertifikātu.
(MK 29.09.2009. noteikumu Nr.1106 redakcijā)
35.2
Saīsināto sertifikātu noformē, ņemot vērā iesniegumā norādītās trešās valsts prasības, ciktāl tās nav pretrunā Pasaules Veselības organizācijas (PVO) vadlīnijām farmaceitisko produktu kvalitātes sertifikācijas shēmai starptautiskajā tirdzniecībā. Saīsinātajā sertifikātā norāda vismaz šādu informāciju:
35.21.
sertifikāta nosaukumu: “Farmaceitiskā produkta sertifikāts” vai “Brīvās tirdzniecības sertifikāts” atbilstoši trešās valsts prasībām;
35.22.
produkta nosaukumu. Zālēm norāda stiprumu (aktīvās(-o)
vielas(-u) daudzumu vienā dozējuma, tilpuma vai masas vienībā) un zāļu formu. Aktīvajai vielai norāda starptautisko nepatentēto nosaukumu (INN) vai, ja tāda nav, ķīmisko nosaukumu. Latvijā reģistrētajām zālēm norāda reģistrācijas numuru, reģistrācijas datumu un derīguma termiņu, ja tāds ir noteikts;
vielas(-u) daudzumu vienā dozējuma, tilpuma vai masas vienībā) un zāļu formu. Aktīvajai vielai norāda starptautisko nepatentēto nosaukumu (INN) vai, ja tāda nav, ķīmisko nosaukumu. Latvijā reģistrētajām zālēm norāda reģistrācijas numuru, reģistrācijas datumu un derīguma termiņu, ja tāds ir noteikts;
35.23.
produkta sastāvu. Aktīvajai vielai norāda dozējumu vienā dozējuma vienībā (iepakojuma vienībā);
35.24.
ražotāja nosaukumu (komersanta firmu), vienoto reģistrācijas numuru Komercreģistrā, juridisko adresi, speciālās atļaujas (licences) (ja tāda ir) nosaukumu, numuru, izdevēju, izdošanas datumu, derīguma termiņu un ražotnes adresi;
35.25.
par zāļu laišanu tirgū atbildīgās personas vārdu, uzvārdu vai firmu un juridisko adresi (attiecas uz Latvijā reģistrētām zālēm);
35.26.
tās personas vārdu, uzvārdu vai firmu un juridisko adresi, uz kuras vārda paredzēts reģistrēt zāles (attiecas uz reģistrācijai iesniegtajām zālēm Latvijā vai citā valstī);
35.27.
ja zāles ir reģistrācijas procesā, to norāda. Ja zāles nav paredzēts reģistrēt, norāda “nav paredzēts reģistrēt laišanai tirgū Latvijā” vai “paredzēts tikai eksportēšanai”. Ja zāles vai aktīvo vielu nav atļauts izplatīt Latvijā, norāda atbilstošo iemeslu, piemēram, “reģistrācija apturēta”, “reģistrācija anulēta” vai “reģistrācija ir atteikta”. Aktīvajai vielai norāda “nav jāreģistrē” vai “paredzēts tikai eksportēšanai”;
35.28.
apliecinājumu, ka attiecīgo produktu var brīvi tirgot konkrētās trešās valsts tirgū saskaņā ar attiecīgās valsts normatīvajos aktos noteiktajām prasībām un pamatojoties uz konkrētā ražotāja (norāda tā nosaukumu) kvalitātes specifikācijām un ka par minētā produkta kvalitāti ir atbildīgs konkrētais ražotājs, kurš ir pakļauts regulārai labas ražošanas prakses pārbaudes un sertifikācijas procedūrai (norāda pārbaužu veikšanas biežumu saskaņā ar normatīvajos aktos par zāļu ražošanu un kontroli noteikto kārtību).
(MK 29.09.2009. noteikumu Nr.1106 redakcijā)
35.3
Saīsinātajā sertifikātā norāda, ka atbilstošā produkta kvalitātes specifikācija ir izstrādāta, pamatojoties uz Eiropas Farmakopejas kvalitātes rādītājiem un aktīvās vielas ražotāja kvalitātes specifikāciju, ja saīsinātā sertifikāta pieprasītājs iesniegumā ir norādījis šādas informācijas nepieciešamību, pamatojot to ar trešās valsts prasībām. Minētā informācija nav jānorāda, ja aktīvai vielai ir Eiropas Zāļu kvalitātes direktorāta izsniegts atbilstības sertifikāts.
35.4
Saīsinātajam sertifikātam pievieno zāļu apraksta, lietošanas instrukcijas un marķējuma kopiju. Minētā prasība neattiecas uz sertifikātu saīsināto formu, kuru izsniedz aktīvajai vielai.
(MK 29.09.2009. noteikumu Nr.1106 redakcijā)
36.
Lai saņemtu šo noteikumu 35.1.apakšpunktā minēto produkta sertifikātu, zāļu ražotājs iesniedz Zāļu valsts aģentūrā iesniegumu, kurā norāda:
36.1.
sertifikāta pieteicēja vārdu, uzvārdu vai firmu un adresi, kā arī kontaktinformāciju (tālruni, faksu un elektroniskā pasta adresi);
36.3.
ja sertifikāta pieteicējs nav zāļu formas ražotājs, norāda zāļu formas ražotāja firmu un adresi;
36.4.
produkta nosaukumu, devu un zāļu formu:
36.4.1.
Latvijā;
36.4.2.
citās valstīs;
36.5.
aktīvo vielu nosaukumu (lietojot starptautisko nepatentēto nosaukumu (INN) vai nacionālo nepatentēto nosaukumu) un to daudzumu vienā devā;
36.6.
pilnu sastāvu, ieskaitot palīgvielas (norāda arī kvantitatīvo sastāvu, ja ir pievienots saskaņojums ar zāļu reģistrācijas īpašnieku);
36.7.
vai produkts ir reģistrēts Latvijā;
36.8.
vai produktu izplata Latvijā;
36.9.
produkta reģistrācijas apliecības numuru un piešķiršanas datumu (ja nepieciešams, norāda, vai reģistrācijas apliecība ir pagaidu vai arī produkts vēl nav apstiprināts);
36.10.
produkta zāļu reģistrācijas īpašnieka nosaukumu un adresi;
36.12.
ja zāļu reģistrācijas īpašnieks nav zāļu formas ražotājs, norāda zāļu formas ražotāja firmu un adresi un pievieno dokumentu, kas apliecina, ka zāļu reģistrācijas īpašnieks piekrīt publiskot šo informāciju;
36.13.
ja reģistrācijas apliecība produktam nav pieprasīta, norāda vienu no iemesliem, kāpēc tā nav nepieciešama:
36.13.1.
produkts radīts tikai speciālas ārstēšanas apstākļiem, galvenokārt tropisku slimību ārstēšanai, kurām nav endēmisks raksturs Latvijā;
36.13.2.
produkts pārstrādāts, lai uzlabotu tā stabilitāti tropiskajos apstākļos;
36.13.3.
produkts pārstrādāts, lai tā sastāvā nebūtu palīgvielu, kuru lietošana importētājvalstī nav atļauta;
36.13.4.
produkts pārstrādāts, lai izveidotu citu maksimāli iespējamo aktīvās vielas devu;
36.13.5.
citi iemesli (norāda, kādi);
36.14.
ja produkta zāļu reģistrācijas īpašnieka vai sertifikāta pieteicēja statuss atbilst šo noteikumu 36.2.2. vai 36.2.3.apakšpunktā minētajam statusam (īpaši, ja produkta ražošanā iesaistīts ārvalstu ražotājs), sertifikāta pieteicējs Zāļu valsts aģentūrā iesniedz informāciju, kurā norāda katras ražošanā iesaistītās puses atbildību attiecībā uz katru ražošanas procesa posmu un galaproduktu, kā arī katras puses veiktās kontroles veidu un apjomu.
(Grozīts ar MK 13.08.2013. noteikumiem Nr.540)
37.
Lai saņemtu šo noteikumu 35.2.apakšpunktā minēto paziņojumu par produkta reģistrācijas statusu, persona iesniedz Zāļu valsts aģentūrā iesniegumu, kurā norāda:
37.1.
sertifikāta pieteicēja vārdu, uzvārdu vai firmu un adresi, kā arī kontaktinformāciju (tālruni, faksu un elektroniskā pasta adresi);
37.2.
importētājvalsti;
37.3.
produkta nosaukumu, devu un zāļu formu, aktīvo vielu nosaukumu (lietojot starptautiskos nepatentētos nosaukumus (INN) vai nacionālos nepatentētos nosaukumus) un to daudzumu vienā devā, reģistrācijas apliecības numuru un piešķiršanas datumu. Ja produkts nav reģistrēts, attiecīgi norāda - "netiek prasīta" vai "nav pieprasīta", vai "ir reģistrācijas procesā", vai "reģistrācija ir atteikta".
(Grozīts ar MK 13.08.2013. noteikumiem Nr.540)
37.1
Lai saņemtu šo noteikumu 35.1 punktā minēto saīsināto sertifikātu, Latvijā reģistrēts zāļu ražotājs iesniedz Zāļu valsts aģentūrā iesniegumu. Iesniegumā norāda:
37.11.
sertifikāta pieteicēja vārdu, uzvārdu vai firmu un adresi, kā arī kontaktinformāciju (tālruni, faksu un elektroniskā pasta adresi);
37.12.
trešo valsti, kompetento iestādi un prasības saīsinātajā sertifikātā norādāmajai informācijai, kā arī trešās valsts prasības labas ražošanas prakses sertifikāta derīgumam, ja tādas ir noteiktas;
37.13.
37.14.
ja saīsinātajā sertifikātā iekļaujama šo noteikumu 35.3 punktā minētā informācija, iesniegumam pievieno produkta kvalitātes specifikāciju. Zālēm iesniedz arī aktīvās(-o) vielas(-u) ražotāja kvalitātes specifikāciju(-as).
(MK 29.09.2009. noteikumu Nr.1106 redakcijā, kas grozīta ar MK 13.08.2013. noteikumiem Nr.540)
38.
Zāļu valsts aģentūra šo noteikumu 35.punktā minēto produkta sertifikātu un paziņojumu par produkta reģistrācijas statusu izsniedz 30 dienu laikā pēc iesnieguma saņemšanas. Izdevumus, kas saistīti ar produkta sertifikāta izsniegšanu, sedz iesnieguma iesniedzējs saskaņā ar Zāļu valsts aģentūras maksas pakalpojumu cenrādi. Zāļu valsts aģentūra sertifikātu un paziņojumu par produkta reģistrācijas statusu izsniedz elektroniska dokumenta formā, nosūtot to uz iesniegumā norādīto elektroniskā pasta adresi. Papīra dokumenta formā sertifikātu un paziņojumu vai tā dublikātu izsniedz pēc pieprasījuma triju darbdienu laikā par papildu maksu saskaņā ar Zāļu valsts aģentūras sniegto maksas pakalpojumu cenrādi.
(Grozīts ar MK 13.08.2013. noteikumiem Nr. 540; MK 02.02.2016. noteikumiem Nr. 82)
38.1
Zāļu valsts aģentūra šo noteikumu 35.1 punktā minēto saīsināto sertifikātu izsniedz 30 dienu laikā pēc iesnieguma saņemšanas. Ja produktam ir izsniegts šo noteikumu 35.1.apakšpunktā minētais produkta sertifikāts un nav nepieciešama atkārtota labas ražošanas prakses atbilstības novērtēšana vai šo noteikumu 35.3 punktā minēto datu novērtēšana, šo noteikumu 35.1 punktā minēto saīsināto sertifikātu Zāļu valsts aģentūra izsniedz 10 dienu laikā pēc iesnieguma saņemšanas. Izdevumus, kas saistīti ar saīsinātā sertifikāta izsniegšanu, saskaņā ar Zāļu valsts aģentūras maksas pakalpojumu cenrādi sedz iesnieguma iesniedzējs.
(MK 29.09.2009. noteikumu Nr. 1106 redakcijā, kas grozīta ar MK 02.02.2016. noteikumiem Nr. 82)
38.2
Zāļu valsts aģentūra saīsināto sertifikātu izsniedz elektroniska dokumenta formā, nosūtot to uz iesniegumā norādīto elektroniskā pasta adresi. Papīra dokumenta formā sertifikātu un paziņojumu vai tā dublikātu izsniedz pēc pieprasījuma triju darbdienu laikā par papildu maksu saskaņā ar Zāļu valsts aģentūras sniegto maksas pakalpojumu cenrādi.
(MK 13.08.2013. noteikumu Nr. 540 redakcijā, kas grozīta ar MK 02.02.2016. noteikumiem Nr. 82)
38.3
Iesniegumu šo noteikumu 35.1.apakšpunktā minētā produkta sertifikāta, šo noteikumu 35.2.apakšpunktā minētā paziņojuma par produkta reģistrācijas statusu un šo noteikumu 35.1 punktā minētā saīsinātā sertifikāta izsniegšanai var iesniegt elektroniska dokumenta formā, to sagatavojot atbilstoši normatīvajiem aktiem par elektronisko dokumentu noformēšanu.
(MK 13.08.2013. noteikumu Nr.540 redakcijā)
39.
Ja šo noteikumu 35.1.apakšpunktā minētā sertifikāta izsniegšanai produktam, kas ir aktīvā viela lietošanai zāļu formā, nepieciešama aktīvās vielas ražošanas vietas pārbaude un aktīvo vielu labas ražošanas prakses atbilstības novērtēšana, sertifikāta pieprasītājs lūdz Zāļu valsts aģentūru veikt aktīvo vielu ražošanas atbilstības novērtēšanu. Minēto pārbaudi veic un labas ražošanas prakses sertifikātu Zāļu valsts aģentūra izsniedz saskaņā ar normatīvajos aktos par zāļu ražošanu un kontroli noteikto kārtību.
(MK 10.06.2008. noteikumu Nr.415 redakcijā)
39.1
Ja šo noteikumu 35.1 punktā minētā saīsinātā sertifikāta izsniegšanai nepieciešama produkta ražošanas vietas pārbaude un labas ražošanas prakses atbilstības novērtēšana, sertifikāta pieprasītājs lūdz Zāļu valsts aģentūru novērtēt produkta labas ražošanas prakses atbilstību un izsniegt labas ražošanas prakses sertifikātu. Zāļu valsts aģentūra minēto pārbaudi veic un labas ražošanas prakses sertifikātu izsniedz saskaņā ar normatīvajos aktos par zāļu ražošanu un kontroli noteikto kārtību.
(MK 29.09.2009. noteikumu Nr.1106 redakcijā)
V.Uzraudzība un sankcijas
40.
Pārtikas un veterinārais dienests:
40.2.
kontrolē zāļu transportēšanas un uzglabāšanas apstākļu atbilstību zāļu uzglabāšanas un transportēšanas prasībām saskaņā ar Eiropas Komisijas publicētajām zāļu labas izplatīšanas prakses pamatnostādnēm (valsts valodā pieejamas Zāļu valsts aģentūras tīmekļvietnē);
40.3.
sniedz Veselības inspekcijai informāciju par šajos noteikumos noteikto prasību pārkāpumiem.
(Grozīts ar MK 10.06.2008. noteikumiem Nr. 415; MK 29.09.2009. noteikumiem Nr. 1106; MK 14.09.2010. noteikumiem Nr. 849; MK 02.02.2016. noteikumiem Nr. 82)
41.
Pārtikas un veterinārais dienests, pamatojoties uz Pārtikas un veterinārā dienesta amatpersonas sastādītu aktu, ir tiesīgs pieņemt lēmumu un apturēt zāļu turpmāku importu:
41.1.
saskaņā ar Eiropas Parlamenta un Padomes regulas Nr. 765/2008 27. pantu, ja konstatē, ka:
41.1.1.
kravas pavaddokumenti neatbilst šo noteikumu 9.punktā noteiktajām prasībām vai zāles nav identificējamas (nav marķējuma);
41.1.2.
importētās zāles nav norādītas Zāļu valsts aģentūras datu bāzē atbilstoši normatīvajiem aktiem par speciālo atļauju (licenču) farmaceitiskajai darbībai izsniegšanas, apturēšanas, pārreģistrēšanas un anulēšanas kārtību;
41.1.4.
ir pārkāptas zāļu uzglabāšanas un transportēšanas prasības saskaņā ar Eiropas Komisijas publicētajām zāļu labas izplatīšanas prakses pamatnostādnēm (valsts valodā pieejamas Zāļu valsts aģentūras tīmekļvietnē);
41.1.5.
zālēm ir beidzies derīguma termiņš;
41.1.6.
zāļu kravas nosūtītājs un saņēmējs nav identificējams;
41.4.
ja komersantam, kurš importē šo noteikumu 41.1.2.apakšpunktā minētās zāles, izsniegtā speciālā atļauja (licence) farmaceitiskajai darbībai nav spēkā;
41.5.
ja uz importētajām zālēm saskaņā ar normatīvajiem aktiem par zāļu izplatīšanas un kvalitātes kontroles kārtību attiecas Veselības inspekcijas ātrās reaģēšanas paziņojums par kvalitātes defektu un zāļu izņemšanu no tirgus;
41.6.
ja ir aizdomas par iespējami viltotām zālēm.
(Grozīts ar MK 10.06.2008. noteikumiem Nr. 415; MK 29.09.2009. noteikumiem Nr. 1106; MK 13.08.2013. noteikumiem Nr. 540; MK 02.02.2016. noteikumiem Nr. 82)
42.
Pārtikas un veterinārais dienests par pieņemto lēmumu paziņo Veselības inspekcijai lēmuma pieņemšanas dienā, bet ne vēlāk kā triju darbdienu laikā un informē inspekciju par vietu, kurā novieto zāles.
43.
(Svītrots)
44.
Ja Pārtikas un veterinārais dienests ir pieņēmis šo noteikumu 41.punktā minēto lēmumu, pēc apstākļu galīgas noskaidrošanas Veselības inspekcija pieņem lēmumu par zāļu importa apturēšanas atsaukšanu vai par zāļu importa aizliegšanu un par pieņemto lēmumu paziņo Pārtikas un veterinārajam dienestam un Zāļu valsts aģentūrai lēmuma pieņemšanas dienā. Ja Veselības inspekcija pieņem lēmumu par importa apturēšanas atsaukšanu, Pārtikas un veterinārais dienests minētā lēmuma saņemšanas dienā informē muitas iestādes, ka ir atļauts piemērot muitas procedūru - laišana brīvā apgrozījumā.
(Grozīts ar MK 10.06.2008. noteikumiem Nr.415; MK 29.09.2009. noteikumiem Nr.1106)
45.
(Svītrots ar MK 02.02.2016. noteikumiem Nr. 82)
46.
Izdevumus, kas saistīti ar konkrētās zāļu kravas iznīcināšanu vai atpakaļizvešanu, sedz persona, uz kuru attiecas Eiropas Parlamenta un Padomes regulas Nr. 765/2008 29. pantā minētais aizliegums zāles laist tirgū (zāļu valdītājs).
(Grozīts ar MK 02.02.2016. noteikumiem Nr. 82)
48.
Zāļu importētājs kontroles laikā sniedz Zāļu valsts aģentūras amatpersonām šādus datus:
48.1.
datus par katras zāļu sērijas kvalitātes kontroli (kas veikta Eiropas Ekonomikas zonas valstī) saskaņā ar zāļu reģistrācijas dokumentāciju;
48.2.
par imunoloģiskajiem preparātiem un no cilvēka asinīm un plazmas iegūtām zālēm - visu kvalificētās personas apstiprinātu kontroles ziņojumu kopijas.
49.
Veselības inspekcija, pamatojoties uz Zāļu valsts aģentūras ziņojumu, ir tiesīga apturēt konkrētu zāļu vai visu importētāja speciālajā atļaujas (licences) lietā norādīto zāļu importu, vai, sadarbībā ar Zāļu valsts aģentūru izvērtējot katru gadījumu atsevišķi, lemt par zāļu importa apturēšanu, ja:
49.3.
zāļu importētājs kontroles laikā neuzrāda šo noteikumu 48.punktā noteiktos datus un dokumentāciju;
49.4.
zāļu importētājs savu darbību veic farmaceitiskās darbības vietā (adresē) un telpās, kas nav norādītas attiecīgajā speciālajā atļaujā (licencē) un iesniegumā šīs licences saņemšanai, kā arī licences lietā;
49.5.
zāļu importētājs importē zāles, kas nav norādītas attiecīgajā iesniegumā speciālās atļaujas (licences) saņemšanai un licences lietā (neattiecas uz zāļu paraugiem un nereģistrētām zālēm);
49.6.
zāļu importētāja rīcībā nav kvalificētas personas, kuras kvalifikācija un profesionālā pieredze atbilst normatīvajos aktos par zāļu ražošanu un kontroles kārtību noteiktajām prasībām;
49.7.
importēto zāļu ražošana vai zāļu aktīvo vielu ražošana neatbilst labas zāļu vai aktīvo vielu ražošanas prakses prasībām;
49.8.
zāles vai zāļu aktīvās vielas ir viltotas.
(Grozīts ar MK 10.06.2008. noteikumiem Nr. 415; MK 02.02.2016. noteikumiem Nr. 82)
50.
Zāļu valsts aģentūra pilda Eiropas Parlamenta un Padomes regulas Nr. 726/2004 19.pantā minētos kompetentās uzraudzības iestādes pienākumus attiecībā uz zālēm, kas saskaņā ar minēto regulu reģistrētas centralizētā reģistrācijas procedūrā un importētas no trešajām valstīm.
51.
Veselības inspekcija:
51.1.
pārrauga, vai zāļu izplatīšana un transportēšana šo noteikumu 2.1 punktā minētajās vietās atbilst šajos noteikumos, normatīvajos aktos par zāļu izplatīšanu un Eiropas Komisijas publicētajās zāļu labas izplatīšanas prakses pamatnostādnēs (valsts valodā pieejamas Zāļu valsts aģentūras tīmekļvietnē) noteiktajām prasībām;
51.2.
pēc muitas noliktavas turētāja (īpašnieka) pieprasījuma pārbauda zāļu uzglabāšanas vietu un sniedz atzinumu muitas noliktavas turētājam (īpašniekam) par tās atbilstību Eiropas Komisijas publicētajās zāļu labas izplatīšanas prakses pamatnostādnēs (valsts valodā pieejamas Zāļu valsts aģentūras tīmekļvietnē) noteiktajām labas izplatīšanas prakses prasībām;
51.3.
ir tiesīga pieprasīt un saņemt no Zāļu valsts aģentūras, Pārtikas un veterinārā dienesta un citām kompetentām valsts iestādēm informāciju, kas saistīta ar šo noteikumu izpildi;
51.4.
sniedz Zāļu valsts aģentūrai, Pārtikas un veterinārajam dienestam un citām kompetentām valsts iestādēm nepieciešamo informāciju;
51.5.
informē Eiropas Komisiju par visiem lēmumiem, kas pieņemti saskaņā ar Padomes regulas Nr. 953/2003 prasību izpildi;
51.6.
informē Eiropas Komisiju par jebkuriem lēmumiem attiecībā uz produktu konfiskāciju vai iznīcināšanu, kas ir pieņemti saskaņā ar Eiropas Parlamenta un Padomes regulu Nr. 816/2006.
(Grozīts ar MK 10.06.2008. noteikumiem Nr. 415; MK 29.09.2009. noteikumiem Nr. 1106; MK 14.09.2010. noteikumiem Nr. 849; MK 02.02.2016. noteikumiem Nr. 82)
52.
Attiecīgo iestāžu amatpersonas neizpauž kontrolētās personas komercnoslēpumus, kas tām kļuvuši zināmi, pildot dienesta pienākumus saskaņā ar šiem noteikumiem.
53.
Veselības inspekcija, Zāļu valsts aģentūra, Pārtikas un veterinārais dienests un muitas iestādes savas kompetences ietvaros nodrošina operatīvu savstarpēju informācijas apmaiņu, kā arī, lai nepieļautu zāļu novirzīšanu nelegālā apritē, sniedz tiesībaizsardzības iestādēm un Veselības ministrijai informāciju par faktiem, kas tām kļuvuši zināmi.
(Grozīts ar MK 10.06.2008. noteikumiem Nr.415; MK 29.09.2009. noteikumiem Nr.1106)
53.1
Veselības inspekcija, Zāļu valsts aģentūra un Pārtikas un veterinārais dienests sadarbojas savas kompetences ietvaros, lai nodrošinātu, ka zāles, kuras ieved un kuras nav paredzēts laist Eiropas Savienības tirgū, nenokļūst apritē, ja ir pamatotas aizdomas, ka tās ir viltotas.
(MK 13.08.2013. noteikumu Nr.540 redakcijā)
53.2
Muitas iestādes nekavējoties ziņo Veselības inspekcijai un Zāļu valsts aģentūrai, ja ir aizdomas par iespējami viltotām zālēm.
(MK 13.08.2013. noteikumu Nr.540 redakcijā)
VI.Noslēguma jautājumi
54.
Atzīt par spēku zaudējušiem Ministru kabineta 2001.gada 27.februāra noteikumus Nr.88 "Zāļu ievešanas, izvešanas un izplatīšanas noteikumi un zāļu lieltirgotavu atvēršanas un darbības prasības" (Latvijas Vēstnesis, 2001, 35., 52.nr.; 2003, 114.nr.; 2004, 69.nr.).
55.
Zāļu lieltirgotavas, kurām uz šo noteikumu spēkā stāšanās dienu ir izsniegta speciāla atļauja (licence) zāļu lieltirgotavas atvēršanai ar speciālās darbības nosacījumu - zāļu ievešana Latvijā no valsts, kas neatrodas Eiropas Ekonomikas zonā, un Zāļu valsts aģentūras izsniegta atļauja attiecīgo zāļu ievešanai Latvijas Republikā no trešajām valstīm, ir tiesīgas importēt zāles līdz normatīvajos aktos, kas nosaka speciālas atļaujas (licences) farmaceitiskajai darbībai izsniegšanas, apturēšanas, pārreģistrēšanas un anulēšanas kārtību, minētās speciālās atļaujas (licences) zāļu ražošanai/importēšanai saņemšanai, bet ne ilgāk kā līdz 2008.gada 1.janvārim.
56.
Sponsors, kuram uz šo noteikumu spēkā stāšanās dienu ir Zāļu valsts aģentūras izsniegta atļauja klīniski pētāmo cilvēkiem paredzēto zāļu ievešanai Latvijas Republikā, ir tiesīgs importēt pētāmās zāles līdz normatīvajos aktos, kas nosaka speciālas atļaujas (licences) farmaceitiskajai darbībai izsniegšanas, apturēšanas, pārreģistrēšanas un anulēšanas kārtību, minētās speciālās atļaujas (licences) zāļu ražošanai/importēšanai saņemšanai, bet ne ilgāk kā līdz 2008.gada 1.janvārim.
56.2
Šo noteikumu 16.1 punktu piemēro ar 2019. gada 9. februāri, ievērojot deleģētās regulas Nr. 2016/161 48. un 50. pantā noteiktos pārejas pasākumus.
(MK 15.01.2019. noteikumu Nr. 39 redakcijā)
56.3
Šo noteikumu 7. punkts ir spēkā līdz 2022. gada 31. decembrim.
56.4
Šo noteikumu 7.2 punkts stājas spēkā 2023. gada 1. janvārī.
56.5
Šo noteikumu 7.2 punktā minētais paziņojums neattiecas uz tām muitas noliktavām, kurām ir izdots Veselības inspekcijas atzinums par noliktavas atbilstību zāļu uzglabāšanas prasībām, un tām muitas noliktavām, kuras norādītas zāļu lieltirgotavas speciālā atļaujā licencē.
56.6
Šo noteikumu 51.2. apakšpunkts ir spēkā līdz 2022. gada 31. decembrim.
56.7
Atļauja zāļu paraugu importam Latvijas Republikā, ko izsniegusi Zāļu valsts aģentūra līdz šo noteikumu spēkā stāšanās dienai, ir derīga.
56.8
Iesniegums atļaujas saņemšanai zāļu paraugu importam Latvijas Republikā , kurš iesniegts Zāļu valsts aģentūrā līdz šo noteikumu spēkā stāšanās dienai, uzskatāms par derīgu.
57.
Noteikumi stājas spēkā ar 2007.gada 1.augustu.
Informatīva atsauce uz Eiropas Savienības direktīvām
(Grozīta ar MK 29.09.2009. noteikumiem Nr.1106; MK 13.08.2013. noteikumiem Nr.540; MK 25.09.2018. noteikumiem Nr. 610; atsauces 7. punkts stājas spēkā 31.01.2022., sk. grozījumu 2. punktu)
Noteikumos iekļautas tiesību normas, kas izriet no:
1)
(svītrots no 31.01.2022. ar MK 25.09.2018. noteikumiem Nr. 610; sk. grozījumu 2. punktu);
2)
Eiropas Parlamenta un Padomes 2001.gada 6.novembra Direktīvas 2001/83/EK par Kopienas kodeksu, kas attiecas uz cilvēkiem paredzētām zālēm;
3)
(svītrots no 31.01.2022. ar MK 25.09.2018. noteikumiem Nr. 610; sk. grozījumu 2. punktu);
4)
Eiropas Parlamenta un Padomes 2004.gada 31.marta Direktīvas 2004/27/EK, ar ko groza Direktīvu 2001/83/EK par Kopienas kodeksu, kas attiecas uz cilvēkiem paredzētām zālēm (dokuments attiecas uz Eiropas Ekonomikas zonu);
5)
(Svītrots)
6)
(svītrots no 31.01.2022. ar MK 25.09.2018. noteikumiem Nr. 610; sk. grozījumu 2. punktu);
7)
Komisijas 2017. gada 15. septembra Direktīvas Nr. 2017/1572, ar ko Eiropas Parlamenta un Padomes Direktīvu 2001/83/EK papildina attiecībā uz cilvēkiem paredzētu zāļu labas ražošanas prakses principiem un pamatnostādnēm.
8)
Eiropas Parlamenta un Padomes 2011.gada 8.jūnija Direktīvas 2011/62/ES, ar ko Direktīvu 2001/83/EK par Kopienas kodeksu, kas attiecas uz cilvēkiem paredzētajām zālēm, groza attiecībā uz to, kā novērst viltotu zāļu nokļūšanu legālās piegādes ķēdē.
1.
pielikumsMinistru kabineta
2007.gada 26.jūnija
noteikumiem Nr.436
Atļauja zāļu paraugu importam Latvijas Republikā
2.
pielikumsMinistru kabineta
2007.gada 26.jūnija
noteikumiem Nr.436
Iesniegums atļaujas saņemšanai zāļu paraugu importam Latvijas Republikā
3.
pielikumsMinistru kabineta
2007.gada 26.jūnija
noteikumiem Nr.436


4.
pielikumsMinistru kabineta
2007.gada 26.jūnija
noteikumiem Nr.436
(Pielikums grozīts ar MK 13.08.2013. noteikumiem Nr.540)

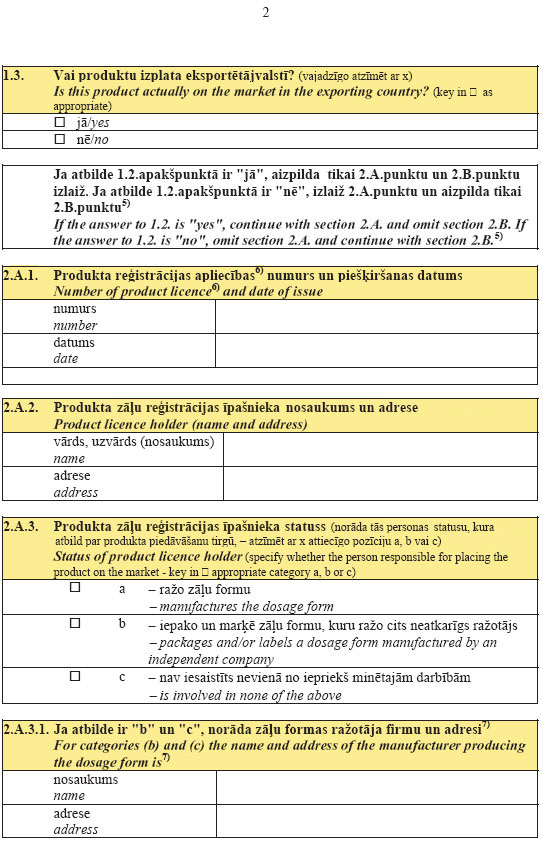
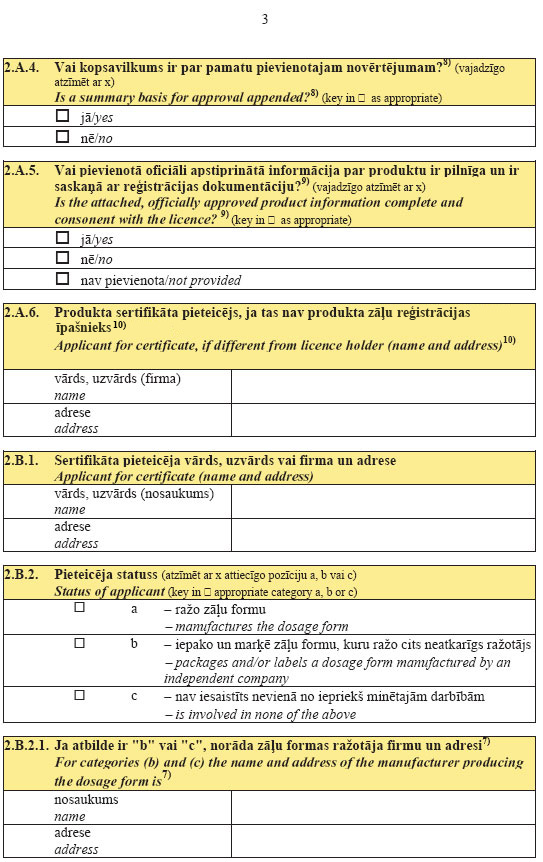
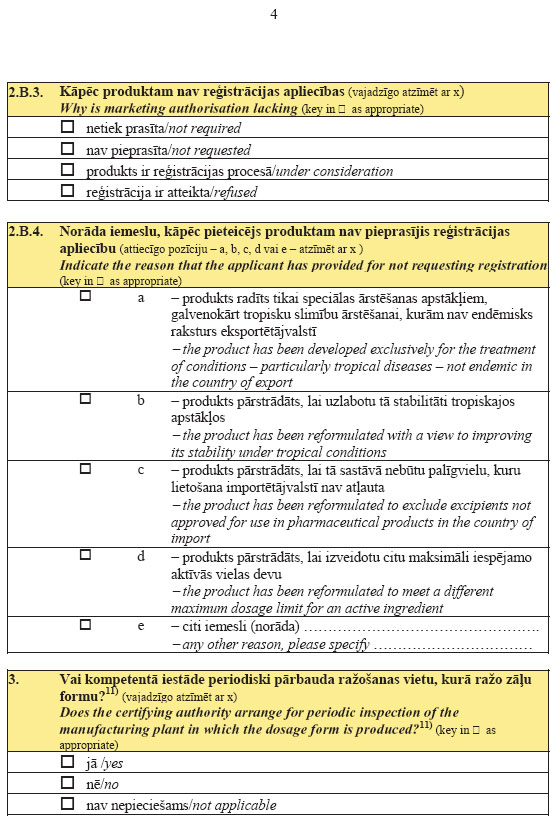



5.
pielikumsMinistru kabineta
2007.gada 26.jūnija
noteikumiem Nr.436
(Pielikums grozīts ar MK 13.08.2013. noteikumiem Nr.540)